- No products in the cart.
Base-modified DNA could potentially find widespread application in medicinal chemistry and therapy, as well as in material science.[1] Examples of applications of base-modified oligonucleotides include aptamers for therapy[2], as scorpion primers[3] or molecular beacons [4] for diagnostic for mutation detection. However, at present, the most important use of base-modified DNA resides in their use in Next Generation Sequencing or Sanger Sequencing.[5]
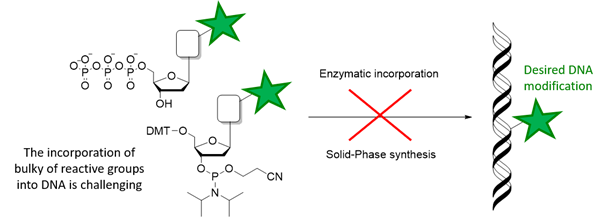
The construction of base-modified oligonucleotides or DNA is, thus, a goal of primary importance. Generally, base-modifications are introduced into DNA by using base-modified nucleotides in enzymatic techniques for DNA synthesis (such as PEX, PCR, NEAR…), or by using base-modified phosphoramidites in the solid-phase chemical synthesis of DNA, using a DNA synthesizer.
However, several limitations are present in these techniques when reactive groups or very bulky groups need to be introduced in the final DNA products. As a matter of fact, DNA polymerases can struggle to introduce very bulky groups and can be inhibited by the presence of chemically reactive groups. On the other hand, the chemical synthesis of oligonucleotide is based on very sentitive phosphorous(III) chemistry, which limits the use of reactive groups (such as azides or nucleophiles).
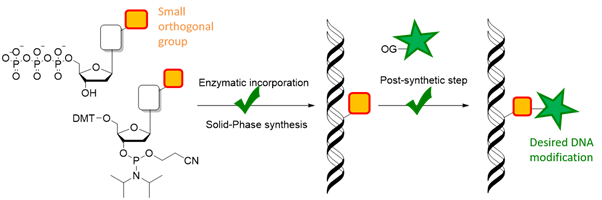
To circumnavigate these inconveniences, a different approach based on a post-synthetic functionalization of DNA has been designed.[6] This approach consists of the enzymatic or chemical incorporation of small reactive groups into oligonucleotides, that are able to undergo a second chemical step directly performed onto the DNA product, to link the final modification to it. Therefore, it is crucial to have in hands building blocks that could readily introduce reactive groups into DNA, which would then efficiently and orthogonally undergo a second synthetic step to yield a final modified product.
CuAAC for oligonucleotide post-synthetic functionalization
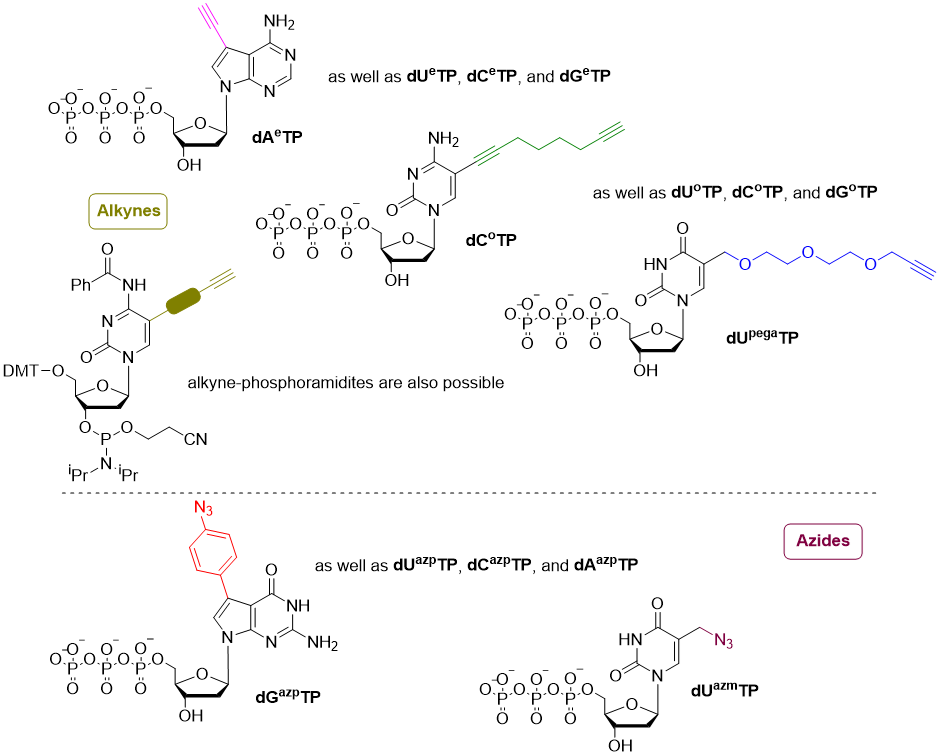
The Copper(I) catalyzed alkyne-azide 1,3-dipolar cycloaddition (CuAAC)[7] is a fast high-yielding 1,3-dipolar cycloaddition reaction, suitable for a post-synthetic modification of oligonucleotides.

The reaction takes place between an alkyne and chemical synthesis, and an azide, both of which can easily be introduced into DNA using azido-modified nucleotide triphosphates and DNA polymerases. Additionally, unlike the azide functionality, alkynes tolerate solid-phase synthesis conditions, thus enabling post-synthetic functionalization of oligonucleotides prepared in this manner.
Some nucleotides for the synthesis of base-modified DNA via post-synthetic CuAAC at SantiagoLab:
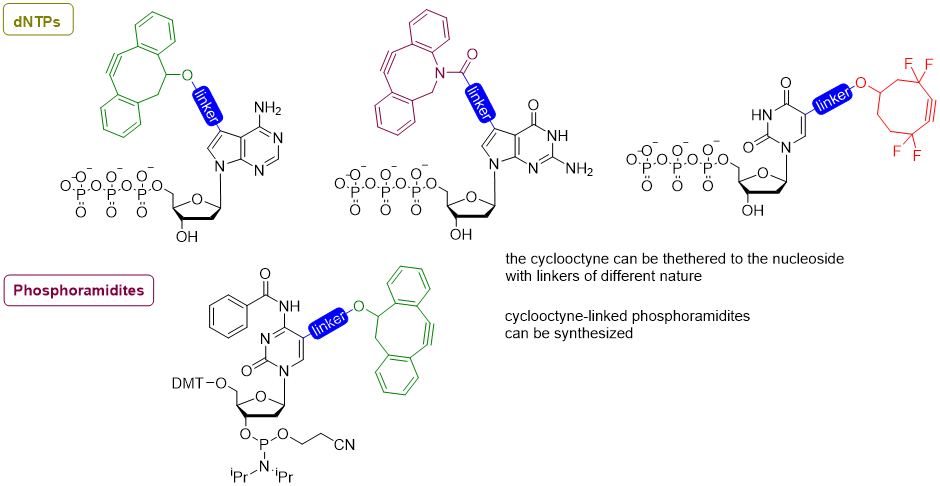
SPAAC for oligonucleotide post-synthetic functionalization
Similarly to the CuAAC, the Strain-Promoted Alkyne-Azyde Cycloaddition (SPAAC) is a reaction between an alkyne and an azide. Differently from the CuAAC, no copper catalyst is needed.[8]
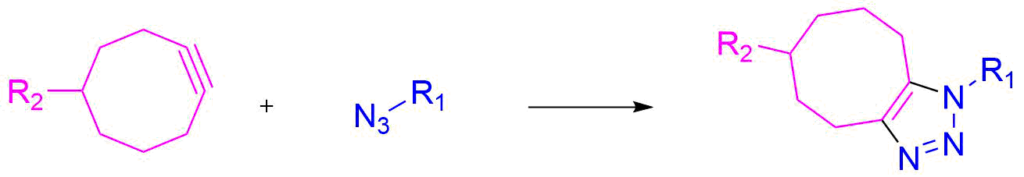
This reaction exploits the high intrinsic reactivity of cyclooctynes, and is a perfect tool for post-synthetic functionalization of oligonucleotides, whereas the absence of the catalyst could be of great advantage when the reaction has to be performed in vivo or in cellulo, where copper cyto-toxicity may be an issue.
Some nucleotides for the synthesis of base-modified DNA via post-synthetic SPAAC @SantiagoLab:
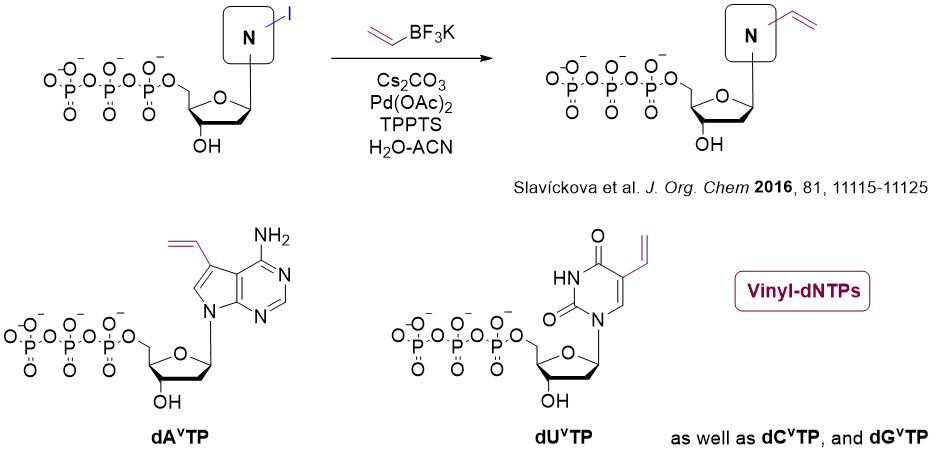
Thiol-ene click
The alkene hydrothiolation (or thiol-ene reaction) takes place between a terminal alkene and a thiol, resulting into a thiolether.

Vinyl nucleotides can be easily synthesized by Suzuki coupling between potassium vinyltrifluoroborate and base-iodinated nucleotides.[9] These vinyl-modified nucleotides are excellent substrates for DNA polymerases.[10]
If you would like to know more about or if we can help you with your research, write an email to Krystof Sigut on krystof.sigut@santiago-lab.com or reach him on the phone +420 776 750 591.
Drop us a line
We will respond as soon as possible, typically within 48 hours.
References:
[1] a) M. Famulok, J. S. Hartig and G. Mayer, Chem. Rev., 2007, 107, 3715–3743; b) G. Mayer, Angew. Chemie Int. Ed., 2009, 48, 2672–2689; c) T. Tørring, N. V. Voigt, J. Nangreave, H. Yan and K. V. Gothelf, Chem. Soc. Rev., 2011, 40, 5636–5646.
[2] Odeh F. et al. Molecules 2020, 25, 3
[3] McKeen C. M. et al. Org. Biomol. Chem., 2003,1, 2267-2275
[4] R. J. Darby et al. Nucleic Acids Res., 2002, 30, e39.
[5] Nature Biotechnology, 2008, 1135–1145.
[6] J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21.
[7] a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chemie Int. Ed., 2001, 40, 2004–2021. b) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015. c) E. Haldón, M. C. Nicasio and P. J. Pérez, Org. Biomol. Chem., 2015, 13, 9528–9550.
[8] a) G. Wittig, R. Pohlke, Chem. Ber. 1961, 94, 3276–3286. b) R. B. Turner, A. D. Jarrett, P. Goebel, B. J. Mallon, J. Am. Chem. Soc. 1973, 95, 790–792.
[9] M. Slavíčkova, R. Pohl and M. Hocek, J. Org. Chem., 2016, 81, 11115-11125.
[10] H. Cahová, A. Panattoni, P. Kielkowski, J. Fanfrlík and M. Hocek, ACS Chem. Biol., 2016, 11, 3165–3171.
