- No products in the cart.
During the course of the last decades, outstanding efforts were devoted to design and develop nucleotide prodrugs to be employed as potential therapeutic agents, especially for the treatment of human immunodeficiency virus (HIV), Ebola, hepatitis B (HBV), hepatitis C (HCV), and herpes simplex virus (HSV). The active form of nucleotide-based drugs is their respective 5’-O-triphosphates (NTPs).
Nucleosides can penetrate cellular membranes. However, once inside the cell, their first phosphorylation to monophosphate (NMP) is hard to accomplish since the NMP-kinases responsible for this step are highly substrate-specific and may not recognize modified analogues. On the other hand, NDP- and NTP-kinases are less specific and are generally able to convert NMP analogues to their relative diphosphates (NDP), and further active NTPs. However, NMPs do not penetrate the cellular membrane, due to their highly polar phosphate group, and their delivery within cells is often very challenging. Phosphatase degradation of NMPs is also an issue.
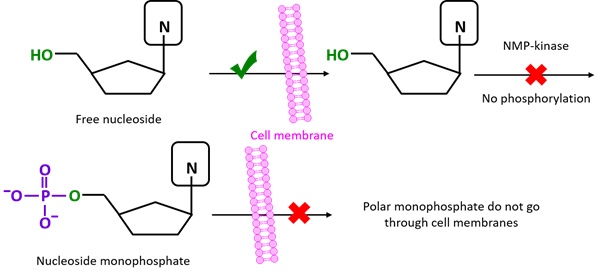
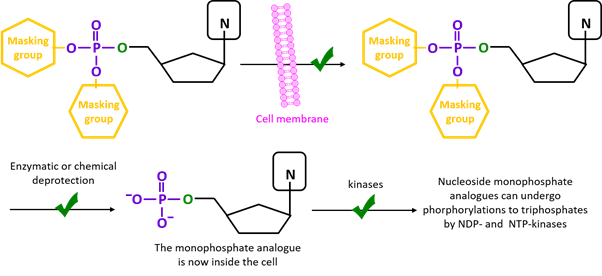
The main idea behind prodrugs consists of masking the phosphate group of NMP analogues with lipophilic groups, thus enabling an efficient delivery across cellular barriers. Once prodrugs reach and penetrates the target cells, the protecting groups are degraded either chemically or enzymatically, and the monophosphate is released.
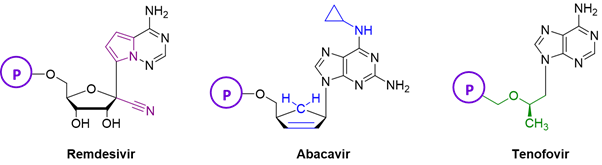
Nucleotide analogues that are currently employed as antiviral agents have structures closely related to natural nucleotides or deoxyribonucleotides. Both sugar and nucleobase modifications are the keys of the activity of nucleotide analogue drugs. Functional group addition or removal, and heteroatom substitutions are the most common modification, as well as the opening of the furanose ring. A few examples are shown in the following figure.
Several types of pro-moieties have been developed and applied to mask phosphate groups of antiviral NMP agents, to improve their delivery and activity. Most often, the choice of enzymatically cleavable phosphate protecting groups is preferred. Several phosphate masking groups have been designed in this field, most of which can be cleaved by esterase enzymes, thanks to their broad substrate specificity. The pivaloyloxymethyl (POM), isopropyloxycarbonyloxymethyl (POC), S-acyl-2-thioethyl (SATE), and 4-acyloxybenzyl (AOB) protecting groups are among the most successfully employed in this class.
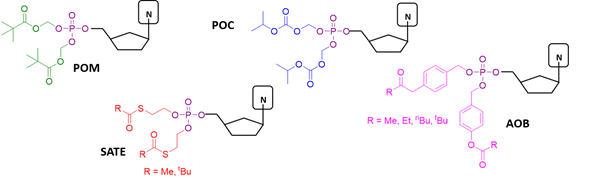
Phosphoramidase-labile protecting groups are another important class of pro-moieties for NMP prodrugs construction. A further class of nucleotide prodrugs does not require the action of any enzyme, and the antiviral agents carry chemically labile protecting groups on the phosphate group. As a main example, cycloSal prodrugs exploit an easily-breakable salicyl ester that readily saponifies inside the cell to start a cascade of chemical reaction leading to the free monophosphate.
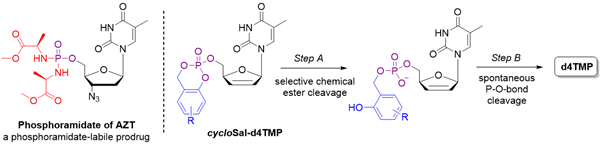
For a comprehensive review on nucleotide, prodrugs see Pradere et al. Chem. Rev. 2014, 9154-9218.
Nucleotide prodrugs @SantiagoLab
@SantiagoLab we gathered vast experience in nucleoside phosphorylation and nucleotide prodrugs synthesis. Initially, we specialized in the synthesis of Remdesivir (for research use only), and you can always find this product in our catalogue. Remdesivir mono-, di- and triphosphates are also available – allowing in vitro studies of the final active antiviral agent.
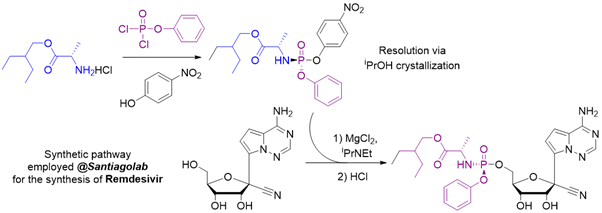
Similarly to Remdesivir, many other nucleotide prodrugs can be synthesized at SantiagoLab.
If you would like to know more about or if we can help you with your research, write an email to Krystof Sigut on krystof.sigut@santiago-lab.com or reach him on the phone +420 776 750 591.
